El término displasia fibrosa (DF) hace referencia a un conjunto de lesiones óseas benignas que se caracterizan por la sustitución del tejido óseo normal por tejido conectivo. En 1891, Von Recklinghausen fue el primero en describir una alteración ósea caracterizada por deformidades y cambios fibrosos que denominó “osteítis fibrosa generalisata”. En 1938, Lichtenstein y Jaffé introdujeron el término “displasia fibrosa”, y reflejaron que esta alteración podía ser monostótica o poliostótica.
La DF es una enfermedad ósea rara de origen no hereditario, causada por una mutación somática en el gen GNAS. La Displasia Fibrosa (DF) es una enfermedad caracterizada por áreas o lesiones del hueso que presentan un crecimiento y una composición anormal. La DF puede existir de manera aislada o asociada a otros trastornos de la piel y del sistema endocrino (hormonal). Los pacientes pueden presentar áreas de la piel hiperpigmentadas denominadas “manchas café con leche”. Los pacientes pueden padecer uno o varios de estos trastornos.
En 1937 McCune y Albright et al relacionaron la variante poliostótica con pubertad temprana y áreas de pigmentación cutánea (manchas café con leche), asociación que en la actualidad se conoce como síndrome de McCune-Allbright y cuya incidencia es de un 4% de los casos de displasia fibrosa.

Manchas café con leche, un signo del síndrome de McCune-Albright.
Tipos de Displasia Fibrosa
- Displasia fibrosa monostótica: Afecta un solo hueso. La variante monostótica supone un 70% del total de casos y en orden de frecuencia afectan a costillas, fémur, tibia, maxilar, mandíbula, calota craneal y húmero.
- Displasia fibrosa poliostótica: Supone aproximadamente un 30% de los casos y clásicamente su comienzo se describe a una edad menor que la variante monostótica. En un 50% de los casos se afecta la región craneofacial. Con mayor frecuencia, y en orden decreciente: maxilar, mandíbula, hueso frontal, hueso esfenoidal, etmoides, hueso parietal y occipital.
La DF craneofacial es una forma muy poco frecuente, que se identifica por lesiones confinadas a huesos contiguos del esqueleto craneofacial, sin afectación de huesos extracraneales y sin alteraciones endocrinas, por lo que no se puede incluir en el resto de formas y queda como una entidad aparte.
La displasia fibrosa osificante es una variante específica que involucra el desarrollo de tejido fibroso que gradualmente se convierte en tejido óseo. La fibrodisplasia osificante progresiva (FOP) es una condición genética extremadamente rara y grave que resulta en la osificación de tejidos blandos como músculos, tendones y ligamentos. Aunque no es una forma clásica de displasia fibrosa, su inclusión es relevante debido a su asociación con alteraciones óseas.
Etiología
Marie et al y Riminucci et al establecieron que la etiología de la displasia fibrosa radica en una mutación activadora somática de la subunidad a de la proteína G de señalización (Gs-a) en células osteoblásticas, con una distribución en mosaico. La sustitución de una cys o his por arg en posición 201 conlleva una pérdida de la actividad guanosina trifosfato de Gs-a. Esto permite la activación de la adenilato ciclasa y, por tanto, la sobreproducción de AMP-c con el consiguiente incremento en la proliferación celular y una diferenciación celular inadecuada.
Dada la complejidad de la DF/MAS, la variabilidad de las manifestaciones entre pacientes y la posibilidad de que distintos órganos se vean afectados, es habitual que se requiera un seguimiento por diferentes especialistas para establecer un manejo óptimo y personalizado.
La etiología de la DF es, muy probablemente, una mutación en el gen Gsa (GNAS1) del cromosoma 20q11. Esta mutación puede ocurrir durante el desarrollo embrionario, o la vida posnatal. La mutación del gen Gsa produce un aumento de la adenilato ciclasa, que aumenta el AMPc intracelular. La alta concentración del AMPc intracelular genera un aumento en la proliferación y diferenciación inapropiada de las células mutadas, causando la formación de una matriz fibrosa inmadura y desorganizada, generando el tejido fibroso de la displasia.
Estas células osteoblásticas también producen interleucina 6 en exceso, con lo que la actividad osteoclástica está incrementada. De esta forma, se producen lesiones osteolíticas expansivas que afectan al tejido fibroso y al hueso normal adyacente.
La DF/MAS es secundaria a una mutación en una de las células del embrión. Esta mutación ocurre después de la concepción y no afecta a todas las células. Si la mutación ocurre en una etapa muy temprana del desarrollo del embrión, afectará a una célula con capacidad de dar lugar a muchos tejidos (célula madre pluripotente). Se desconoce qué causa la mutación de la DF/MAS. Al parecer, el gen implicado es especialmente susceptible a alterarse, es decir, propenso a las mutaciones. Aunque la DF se considere una enfermedad rara, afecta a personas de ambos sexos y de todas las razas y lugares del mundo, por lo que se considera que la mutación ocurre de manera aleatoria.
Características histológicas
Histológicamente la displasia fibrosa se caracteriza por una extensa proliferación de tejido fibroso que se entremezcla de forma irregular con trabéculas de hueso inmaduro. Se han descrito tres patrones diferentes por los que estas trabéculas se pueden agrupar: “letras chinas”; “puzzle” y “C&S”. Además, la matriz osteoide contiene osteoblastos inmaduros dispersos de modo irregular.
Histológicamente muestra trabéculas de hueso inmaduro en el seno de un estroma fibrocelular en el cual se pueden encontrar osteoclastos dispersos. Las trabéculas son delgadas y gráciles, no estando conectadas unas con otras y muestran una morfología curvilínea, con patrón de "letras chinas", "puzzle" o en "C&S".
Microfotografía de displasia fibrosa mostrando el patrón de "letras chinas".
Manifestaciones clínicas
La clínica relacionada con las lesiones de displasia fibrosa va a depender en gran medida de la localización y del tamaño de la lesión. De esta forma, podemos encontrarnos con lesiones de relativo gran tamaño prácticamente asintomáticas hasta lesiones de escaso volumen que, dada su localización (generalmente la región craneofacial), pueden producir alteraciones funcionales (por ejemplo, ceguera en lesiones del conducto óptico) o estéticas importantes.
La clínica más frecuentemente observada es la derivada del agrandamiento gradual sin dolor del hueso implicado, en este caso, la asimetría facial, con su correspondiente deformidad estética. Otros síntomas son resultado de la constricción de agujeros craneales o de la obliteración de cavidades óseas: anosmia, diplopia, proptosis, epífora, estrabismo, parálisis facial, tinnitus, obstrucción nasal, maloclusión e interferencia con la masticación y el habla.
Los síntomas de la displasia fibrosa dependen del número y localización de los huesos afectados, así como del grado de compromiso óseo. Los pacientes pueden presentar áreas de la piel hiperpigmentadas denominadas “manchas café con leche”. Los pacientes pueden padecer uno o varios de estos trastornos.
Dado que la DF craneofacial generalmente no altera las estructuras vitales del cráneo, incluyendo los vasos sanguíneos, el cerebro o las neuronas, cuando un paciente con DF sufre dolor, el primer paso es descartar causas ortopédicas, tales como fracturas, quistes del hueso, y problemas de la marcha. A los pacientes también se les debe revisar los niveles de fosfato, ya que niveles bajos pueden empeorar el dolor óseo.
Las lesiones de DF suelen manifestarse durante los primeros años de vida y tienden a aumentar de tamaño durante la infancia. La gran mayoría de las lesiones se pueden detectar antes de los 10 años de edad, siendo muy raro que aparezcan nuevas lesiones después de los 15 años.
La DF puede afectar un hueso (monostótica) o varios huesos (poliostótica), siendo más común su manifestación en la infancia o al inicio de la adolescencia. La DF poliostótica puede relacionase con pubertad temprana y áreas de pigmentación cutánea.
Diagnóstico
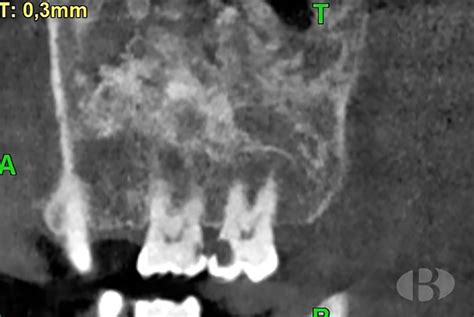
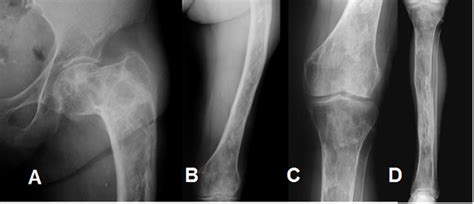
Desde un punto de vista radiográfico, las lesiones de displasia fibrosa se caracterizan por un aspecto de vidrio deslustrado debido a la mezcla de elementos óseos y fibrosos. La densidad radiográfica de la lesión dependerá de la proporción relativa de estos elementos. Así pueden adoptar un patrón esclerótico, quístico (lítico) o mixto. La variante esclerótica constituye un 35% de los casos descritos y tiende a localizarse en la base del cráneo. La variante mixta es la más frecuente (40% de los casos), y el patrón quístico es el menos frecuente.
Por ello es necesario un diagnóstico, además de clínico, basado en la imagen radiográfica (ya casi no utilizada) y más frecuentemente de TC, donde se describen varias formas (Schumberger, 1946):
- Pagetoide: expansión ósea con islas dispersas de formación ósea en campo de baja atenuación
- Esclerótica: apariencia homogénea con patrón en "vidrio esmerilado"
- Cística: lesión bien definida de baja atenuación con borde esclerótico en patrón de "cáscara de huevo"
Ante una lesión diagnosticada de DF por biopsia, será necesario un estudio analítico para descartar alteración endocrina y un TC full body/gammagrafía para descartar una extensión extracraneal. Hay autores que consideran que la medición de la fosfatasa alcalina sérica (FAS), junto con la edad del paciente, pueden ser un buen marcador para detectar la progresión de enfermedad en la cirugía incompleta de la DF craneofacial.
III Congreso ADF - Cómo y cuándo operar la DF en huesos largos - Dra Ana María Bueno Sánchez
Los análisis de sangre son fundamentales para evaluar los niveles de fosfato, calcio, hormonas tiroideas, hormona de crecimiento, y en ocasiones hormonas sexuales.
TC mostrando displasia fibrosa con apariencia de vidrio esmerilado.
Diagnóstico Diferencial
A pesar de que el patrón anatomopatológico es característico, se debe realizar el diagnóstico diferencial con múltiples entidades. El fibroma osificante es una verdadera neoplasia consistente en una proliferación de un tejido fibroso celular, que aparece bien delimitada o raramente encapsulada, que incluye hueso neoformado o calcificaciones esféricas.
El tumor solo se presenta en los maxilares y huesos craneofaciales y tiene su inicio en tercera y cuarta década de la vida. Histológicamente, el componente fibroso muestra fibroblastos que no presentan atipia, escasas células multinucleadas gigantes, y ocasionales macrófagos espumosos. Las trabéculas óseas son finas, curvilíneas y aparecen inconexas, no hallándose bordeadas por un ribete de osteoblastos. Las características histológicas son similares a las de la DF por lo que la distinción hay que hacerla en base a los criterios clínicos, radiológicos, hallazgos quirúrgicos y características.
El querubismo es una enfermedad benigna, autosómica dominante, que consiste en una lesión de células gigantes de la mandíbula/maxilar que se presenta en niños entre 2-5 años como una tumefacción bilateral indolora, con progresión hasta la pubertad, cuando regresa de manera espontánea. Otras entidades que pueden confundirse clínicamente con la DF son el granuloma de células gigantes, el quiste óseo aneurismático, el cementoma gigante, el ameloblastoma o la displasia ósea, entre otros.
Con relación al maxilar, las principales entidades que hay que tener en cuenta junto a la DF son el FO (y su variante juvenil, de rápido crecimiento y carácter destructivo) y el CM, que suelen presentarse entre la tercera y cuarta décadas; además su localización preferente es la mandibular. Cabe decir que la distinción radiológica e incluso anatomopatológica es difícil. De hecho, se ha sugerido que todas estas lesiones osteofibrosas benignas constituyen diferentes estadios evolutivos de una misma entidad.
Tratamiento
El tratamiento de la displasia fibrosa varía según la severidad de los síntomas, la localización de la enfermedad y las complicaciones asociadas. Actualmente, no existe una cura definitiva para la displasia fibrosa. El manejo está orientado a aliviar los síntomas, prevenir complicaciones y mejorar la calidad de vida del paciente. El seguimiento médico regular es esencial para monitorizar la progresión de la displasia fibrosa, especialmente en niños y adolescentes, donde las deformidades pueden empeorar con el crecimiento.
El tratamiento de la DF está en continua revisión y no existen pautas de actuación bien establecidas. En nuestra opinión, el tratamiento de las lesiones del área craneofacial debe basarse en la clínica, excepto en lesiones de cono orbitario, que dado el alto riesgo de compresión del nervio orbitario la indicación está fuera de toda duda. Es aceptable hacer observación cuando las lesiones son pequeñas, asintomáticas y cosméticamente aceptables.
Exceptuando algunos casos de DF monostótica tributarios a la extirpación completa de la lesión por cirugía, no existe en la actualidad una cura definitiva para la DF/MAS.
El tratamiento debe ser conservador y tener como meta prevenir problemas funcionales, conservar la estética y en casos necesarios, la descompresión de nervios craneales. Puede manifestarse en diversas formas clínicas, según el hueso afectado.
Tratamiento Médico
Respecto al tratamiento médico, históricamente la radioterapia (RT) ha sido una de las opciones a considerar, pero actualmente ha sido abandonada por el riesgo de transformación maligna posterior (sarcoma post RT). La quimioterapia (QT) ha demostrado no ser efectiva.
Actualmente se plantea la terapia con bisfosfonatos (como pamidronato o alendronato), con la coadyuvancia del calcio, la vitamina D y el fósforo; sin embargo, existen contradicciones respecto a esta opción terapéutica: por una parte, esta terapia estabiliza la enfermedad, disminuye el riesgo de fracturas óseas y mejora el dolor del paciente. Además, se ha observado un incremento de la actividad de los osteoclastos, muy disminuida en la DF y una mejoría radiológica; sin embargo y a pesar de estos resultados, autores como Plotkin et al. no han demostrado evidencia de mejoría radiológica en sus estudios, a pesar de obtener resultados estadísticamente significativos respecto a los niveles de los marcadores óseos del turnover. Además, el tratamiento con bisfosfonatos no esta claramente indicado en pacientes sin dolor óseo ni con lesiones osteolíticas significativas.
Si el dolor no está relacionado con estos factores, los primeros pasos en el tratamiento deben ser medidas conservadoras, incluyendo reposo, masaje, aplicación de frio y calor, y la ingesta de medicamentos como el ibuprofeno y el paracetamol. Para el dolor moderado o severo que no responde a estos tratamientos, los bifosfonatos intravenosos como el zolendronato y el pamidronato pueden ser útiles. Éstos deben ser administrados en la dosis y la frecuencia más bajas necesarias para controlar el dolor.
Existen medicamentos que pueden disminuir el dolor en huesos, como son los bifosfonatos, los cuales son medicamentos ampliamente usados para la osteoporosis. Estos medicamentos han resultado efectivos en algunos pacientes, pero no en todos.
Alternativamente, la existencia de un incremento en la reabsorción ósea y en la remodelación, ha alentado el desarrollo de algunos ensayos clínicos con calcitonina, para inhibir la resorción osteoclástica. En algunos estudios se observó una disminución de la excreción urinaria de hidroxiprolina y otros encontraron cambios en los niveles de fosfatasa alcalina sérica tras la administración de calcitonina en pacientes con DF. Sin embargo, otros estudios no encontraron cambios en los niveles de fosfatasa alcalina sérica y no se mencionan cambios en la clínica ni radiológicos.
Tratamiento Quirúrgico
Algunos autores realizan remodelado quirúrgico (citorreducción o reducción del tejido lesionado). Sin embargo, en esta opción de tratamiento puede quedar enfermedad residual que siga creciendo y requiera nuevamente cirugía; además, a pesar de ser menos invasivo, el tejido remanente no es adecuado para colocación de implantes dentales.
La escisión radical y reconstrucción (propuesta por primera vez en 1990 por Chen y Noorhoff), es el tratamiento más utilizado en la actualidad. Sus indicaciones son: compresión del nervio óptico, deformidad cosmética, dolor, fractura patológica, disminución de la audición y obstrucción nasal o sinusal. El injerto libre de peroné fue la cirugía propuesta por Taylor et al. en 1975, y posteriormente introducido el método por Hidalgo.
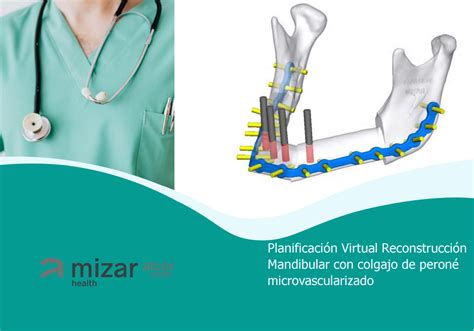
Reconstrucción mandibular con injerto de peroné.
Transformación Maligna
La transformación maligna en la displasia fibrosa es poco frecuente, y las tasas de malignización son de un 0,5% para la variante monostótica y del 4% para la variante poliostótica asociada al síndrome de McCune-Allbright.
Se produce transformación maligna a sarcoma en un porcentaje muy bajo de pacientes (0,4% en DF monostótica y hasta un 4% en McCune Albright). La región craneofacial es el lugar de malignización más frecuente, con 35,6% de conversión (vs. en el fémur que tiene una frecuencia del 24,7% o en la tibia con frecuencia del 12,8%). A pesar de la creencia de que factor de riesgo más importante para que ocurra es la radiación previa, no es estrictamente necesaria, como demostraron Schwartz y Alpert en 1964.
La degeneración neoplásica es muy infrecuente (0,5%) y, curiosamente, en un tercio de los pacientes se han descrito antecedentes de radiación. Puede ocurrir en ambas formas, mono y poliostótica, siendo las transformaciones más frecuentes a osteosarcoma, fibrosarcoma y condrosarcoma, en orden decreciente. Debe sospecharse ante un incremento del dolor o de la tumoración o ante un cambio radiológico significativo respecto a densidad y/o extensión de la tumoración.
Displasia Fibrosa y Embarazo
Muchas mujeres con DF/MAS tienen embarazos exitosos. No hay evidencia de que el embarazo haga que la DF empeore o aumente el dolor óseo. Las mujeres con DF en la columna, las costillas o la pelvis deben realizar un seguimiento con un obstetra especializado en embarazos de alto riesgo. Las mujeres que tienen antecedentes de pubertad precoz continúan teniendo quistes ováricos en la edad adulta. Esto puede derivar en sangrados irregulares e intensos. Las mujeres pueden tardar más en quedarse embarazadas.
| Característica | Descripción |
|---|---|
| Definición | Reemplazo de hueso normal por tejido fibroso anormal. |
| Tipos | Monostótica (un hueso) y poliostótica (múltiples huesos). |
| Etiología | Mutación somática en el gen GNAS. |
| Manifestaciones Clínicas | Deformidades esqueléticas, dolor, fracturas, alteraciones craneofaciales. |
| Diagnóstico | Radiografías, TC, biopsia. |
| Tratamiento | Manejo del dolor, bisfosfonatos, cirugía reconstructiva. |
| Transformación Maligna | Rara, pero posible (osteosarcoma, fibrosarcoma). |