El término displasia fibrosa (DF) hace referencia a un conjunto de lesiones óseas benignas que se caracterizan por la sustitución del tejido óseo normal por tejido conectivo. La DF es una enfermedad ósea rara que puede afectar uno o varios huesos del cuerpo. Aunque la DF se considere una enfermedad rara, afecta a personas de ambos sexos y de todas las razas y lugares del mundo, por lo que se considera que la mutación ocurre de manera aleatoria.
En 1891, Von Recklinghausen fue el primero en describir una alteración ósea caracterizada por deformidades y cambios fibrosos que denominó “osteítis fibrosa generalisata”1. En 1938, Lichtenstein y Jaffé2 introdujeron el término “displasia fibrosa”, y reflejaron que esta alteración podía ser monostótica o poliostótica. En 1937 McCune3 y Albright et al4 relacionaron la variante poliostótica con pubertad temprana y áreas de pigmentación cutánea (manchas café con leche), asociación que en la actualidad se conoce como síndrome de McCune-Allbright y cuya incidencia es de un 4% de los casos de displasia fibrosa.
La DF puede existir de manera aislada o asociada a otros trastornos de la piel y del sistema endocrino (hormonal). Los pacientes pueden presentar áreas de la piel hiperpigmentadas denominadas “manchas café con leche”. Los pacientes pueden padecer uno o varios de estos trastornos. La DF/MAS es secundaria a una mutación en una de las células del embrión. Esta mutación ocurre después de la concepción y no afecta a todas las células. Si la mutación ocurre en una etapa muy temprana del desarrollo del embrión, afectará a una célula con capacidad de dar lugar a muchos tejidos (célula madre pluripotente). Se desconoce qué causa la mutación de la DF/MAS. Al parecer, el gen implicado es especialmente susceptible a alterarse, es decir, propenso a las mutaciones.
La DF se considera una lesión benigna caracterizada por la sustitución de tejido óseo normal por tejido fibro-óseo conectivo. Descrita por primera vez por von Recklinghausen en 1981, el término «displasia fibrosa» fue establecido por Lichtenstein y Jaffe en 1938, describiendo a su vez 2 tipos de DF: monostótica (DFM) y poliostótica (DFP) (afectación de 2 o más regiones óseas).
Este es un trastorno óseo no hereditario que ocurre cuando un error genético provoca el crecimiento desorganizado del hueso. En lugar de desarrollar un hueso normal y fuerte, el cuerpo forma un tejido fibroso inmaduro, lo que da lugar a debilidad estructural.
III Congreso ADF - Cómo y cuándo operar la DF en huesos largos - Dra Ana María Bueno Sánchez
Tipos de Displasia Fibrosa
Esta enfermedad puede presentarse en dos formas principales:
- Displasia fibrosa monostótica: Afecta a un solo hueso y es la forma más común. La variante monostótica supone un 70% del total de casos y en orden de frecuencia afectan a costillas, fémur, tibia, maxilar, mandíbula, calota craneal y húmero.
- Displasia fibrosa poliostótica: Afecta a múltiples huesos y, en algunos casos, puede estar asociada con el síndrome de McCune-Albright, una condición rara que también incluye alteraciones hormonales y manchas en la piel (café con leche). La variante poliostótica supone aproximadamente un 30% de los casos y clásicamente su comienzo se describe a una edad menor que la variante monostótica. En un 50% de los casos se afecta la región craneofacial. Con mayor frecuencia, y en orden decreciente: maxilar, mandíbula, hueso frontal, hueso esfenoidal, etmoides, hueso parietal y occipital5.
Displasia fibrosa del fémur izquierdo.
Causas y Mecanismos de la Enfermedad
La displasia fibrosa es causada por una mutación en el gen GNAS1, que codifica una proteína llamada Gsα. Esta proteína participa en la regulación de múltiples funciones celulares, incluyendo la formación ósea.
Marie et al6 y Riminucci et al7 establecieron que la etiología de la displasia fibrosa radica en una mutación activadora somática de la subunidad a de la proteína G de señalización (Gs-a) en células osteoblásticas, con una distribución en mosaico. La sustitución de una cys o his por arg en posición 201 conlleva una pérdida de la actividad guanosina trifosfato de Gs-a. Esto permite la activación de la adenilato ciclasa y, por tanto, la sobreproducción de AMP-c con el consiguiente incremento en la proliferación celular y una diferenciación celular inadecuada.
Estas células osteoblásticas también producen interleucina 6 en exceso, con lo que la actividad osteoclástica está incrementada. De esta forma, se producen lesiones osteolíticas expansivas que afectan al tejido fibroso y al hueso normal adyacente8.
La mutación ocurre de manera esporádica durante el desarrollo fetal y no se hereda de los padres. Como resultado, las células óseas afectadas proliferan de manera anormal y generan un hueso menos resistente.
El impacto de la mutación depende del número de células afectadas y de la localización de la displasia en el esqueleto. En algunos casos, la enfermedad permanece estable durante años, mientras que en otros puede empeorar con el crecimiento.
Síntomas y Manifestaciones Clínicas
Los síntomas de la displasia fibrosa varían según la severidad y los huesos afectados. Algunos pacientes pueden ser asintomáticos, mientras que otros presentan síntomas significativos.
La clínica relacionada con las lesiones de displasia fibrosa va a depender en gran medida de la localización y del tamaño de la lesión. De esta forma, podemos encontrarnos con lesiones de relativo gran tamaño prácticamente asintomáticas hasta lesiones de escaso volumen que, dada su localización (generalmente la región craneofacial), pueden producir alteraciones funcionales (por ejemplo, ceguera en lesiones del conducto óptico) o estéticas importantes.
Síntomas más comunes:
- Dolor óseo: Generalmente es leve, pero puede empeorar con el tiempo. Siento dolor en los huesos afectados con DF, pero mi médico me ha dicho que la DF no duele. Desafortunadamente, algunos textos de medicina anticuados exponen que las lesiones de DF son indoloras. Si bien hay muchos pacientes con DF que no sienten dolor, hay otros muchos que sí. Parece que el dolor es menos frecuente en niños que en adultos, pero igualmente pueden presentar dolor.
- Deformidades óseas: Los huesos afectados pueden volverse curvados o engrosados, especialmente en los huesos largos (fémur, tibia, húmero).
- Fracturas espontáneas: Debido a la debilidad estructural del hueso, las fracturas pueden ocurrir con traumatismos mínimos.
- Asimetría facial: Si la displasia afecta el cráneo o la mandíbula, puede provocar un crecimiento anormal y cambios en la apariencia.
- Compresión nerviosa: En casos en los que la displasia afecta la base del cráneo o la columna, puede comprimir nervios y causar síntomas neurológicos, como pérdida de la visión o audición.
En la forma poliostótica, los pacientes pueden tener síntomas adicionales debido a alteraciones hormonales, como pubertad precoz en niñas o disfunciones tiroideas.
Diagnóstico
Desde un punto de vista radiográfico, las lesiones de displasia fibrosa se caracterizan por un aspecto de vidrio deslustrado debido a la mezcla de elementos óseos y fibrosos. La densidad radiográfica de la lesión dependerá de la proporción relativa de estos elementos. Así pueden adoptar un patrón esclerótico, quístico (lítico) o mixto. La variante esclerótica constituye un 35% de los casos descritos y tiende a localizarse en la base del cráneo. La variante mixta es la más frecuente (40% de los casos), y el patrón quístico es el menos frecuente.
Histológicamente la displasia fibrosa se caracteriza por una extensa proliferación de tejido fibroso que se entremezcla de forma irregular con trabéculas de hueso inmaduro. Se han descrito tres patrones diferentes por los que estas trabéculas se pueden agrupar: “letras chinas”; “puzzle” y “C&S”. Además, la matriz osteoide contiene osteoblastos inmaduros dispersos de modo irregular.
El elemento crítico para el diagnóstico es la presencia de unos márgenes indefinidos, debido a la sutil mezcla entre hueso lesional y hueso aparentemente normal6. Esta es una característica que permite diferenciar a la DFM del fibroma osificante juvenil (FOJ), que presenta frecuentemente márgenes bien delimitados.
Los análisis de sangre son fundamentales para evaluar los niveles de fosfato, calcio, hormonas tiroideas, hormona de crecimiento, y en ocasiones hormonas sexuales.
¿Existe Tratamiento?
Actualmente, no existe una cura definitiva para la displasia fibrosa, pero el tratamiento se centra en el control de los síntomas y la prevención de complicaciones.
Manejo Médico
El tratamiento médico se basa en el control del dolor y la prevención de la progresión de la enfermedad:
- Analgésicos y antiinflamatorios: Medicamentos como paracetamol o AINEs (ibuprofeno, naproxeno) pueden aliviar el dolor óseo.
- Bifosfonatos: Fármacos como el alendronato pueden ayudar a fortalecer el hueso y reducir el dolor, aunque su eficacia es variable. En términos generales, el tratamiento clásico de las lesiones de displasia fibrosa que producían sintomatología de importancia o alteraciones estéticas significativas implicaba, en la mayor parte de casos, realizar intervenciones quirúrgicas resectivas de mayor o menor amplitud en conjunción con técnicas reconstructivas, especialmente en el ámbito de la cirugía craneofacial11,12. Por el contrario, en los últimos 10 años, se han publicado diversos estudios en los que se analizaba la efectividad del tratamiento con bisfosfonatos para la displasia fibrosa13-15. No obstante, aunque los resultados son prometedores en términos de mejora de la sintomatología y de la densitometría de las lesiones óseas, el número de pacientes incluidos en cada estudio es bajo y, además, no han sido claramente elucidados aspectos como la disminución del dolor.
- Monitoreo endocrinológico: En pacientes con displasia poliostótica, es importante evaluar y tratar cualquier alteración hormonal.
Para el dolor moderado o severo que no responde a estos tratamientos, los bifosfonatos intravenosos como el zolendronato y el pamidronato pueden ser útiles. Éstos deben ser administrados en la dosis y la frecuencia más bajas necesarias para controlar el dolor.
Tratamiento Quirúrgico
En casos donde la enfermedad causa deformidades severas o fracturas repetidas, puede ser necesario recurrir a la cirugía:
- Osteotomías correctivas: Se realizan cortes en el hueso para mejorar la alineación y la función.
- Fijación interna: Se utilizan placas, tornillos o clavos para reforzar el hueso afectado.
- Injertos óseos: En algunos casos, se pueden utilizar injertos de hueso para mejorar la resistencia del área afectada.
La cirugía se indica cuando hay un alto riesgo de fracturas, dolor intenso o deformidades progresivas.
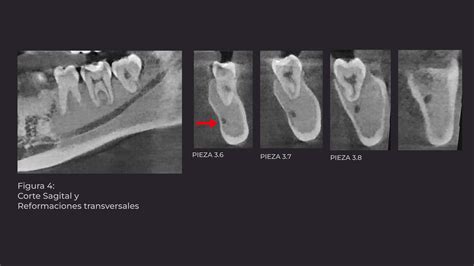
Imagen intraoperatoria de una cirugía de mandíbula.
Pronóstico y Calidad de Vida
El pronóstico de la displasia fibrosa varía según la extensión de la enfermedad. En muchos casos, los pacientes pueden llevar una vida normal con un manejo adecuado del dolor y seguimiento médico. Sin embargo, en formas más graves, puede ser necesario tratamiento quirúrgico para evitar complicaciones a largo plazo.
Es importante el seguimiento regular con un equipo multidisciplinario que incluya ortopedistas, endocrinólogos y especialistas en rehabilitación.
Aunque no tiene cura, existen múltiples estrategias para controlar los síntomas y mejorar la calidad de vida de los pacientes. Un diagnóstico temprano y un manejo adecuado pueden marcar la diferencia en la evolución de la enfermedad.
Si presentas dolor óseo persistente o alguna anomalía en el crecimiento óseo, es fundamental acudir a un especialista para una evaluación adecuada.
Lo Que Debes Saber…
La displasia fibrosa es una enfermedad ósea rara que puede afectar uno o varios huesos del cuerpo. Se caracteriza por el reemplazo anormal del tejido óseo normal por un tejido fibroso, lo que puede debilitar los huesos y provocar deformidades.
En casos donde la enfermedad causa deformidades severas o fracturas repetidas, puede ser necesario recurrir a la cirugía.
El pronóstico de la displasia fibrosa varía según la extensión de la enfermedad.
Conceptos erróneos sobre la Displasia Fibrosa
Mi médico se ha referido a la Displasia Fibrosa como tumor del hueso. La DF NO es cáncer. Las palabras “tumor” o “neoplasia” simplemente indican un crecimiento celular anormal. Los tumores pueden ser malignos (cancerosos) o benignos (no-cancerosos). Para evitar confusiones, algunos médicos prefieren utilizar el término “lesión” en lugar de tumor. Por normal general, se necesitan muchas mutaciones en una misma célula para que se genere un tumor maligno (cáncer). La mutación causante de la DF/MAS se ha encontrado en algunos tumores malignos de la glándula pituitaria (o hipófisis) y menos frecuentemente, en otros tipos de cáncer. Como la mayoría de pacientes con DF/MAS no desarrollan estos tumores malignos, no parece que la mutación de la DF de lugar a cáncer por sí misma.
El médico me ha dicho que la Displasia Fibrosa mejorará después de la adolescencia. Históricamente se consideraba que las lesiones de DF dejaban de crecer al finalizar el crecimiento (al alcanzar la estatura final).
No hay tratamiento que pueda retrasar el crecimiento de las lesiones óseas en la DF. Los bifosfonatos pueden ser útiles para el dolor óseo; sin embargo, no hay pruebas de que hagan efecto sobre el tamaño ni la evolución de las lesiones óseas. Las lesiones de DF suelen manifestarse durante los primeros años de vida y tienden a aumentar de tamaño durante la infancia. La gran mayoría de las lesiones se pueden detectar antes de los 10 años de edad, siendo muy raro que aparezcan nuevas lesiones después de los 15 años (Hart et al 2007).
Impacto en la Calidad de Vida
No hay una respuesta sencilla para esta pregunta. Los efectos difieren enormemente según la extensión de la enfermedad y del paciente. Para algunas personas no supone ningún tipo de impacto, mientras que para otras puede dar lugar a discapacidades tales como requerir una silla de ruedas para moverse, perder capacidad visual, auditiva etc.
Investigaciones Actuales
- “Anti-resorptive drugs in fibrous dysplasia of bone: Studies on the effects of a RANKL inhibitor and Zoledronic Acid in a murine model of the disease by radiography, histology, and genome-wide expression analysis (NanoString)”, Dra.
- “Mechanistic and Therapeutic Studies of Fibrous Dysplasia in a New Mouse Model.” Dr.
- “Identification and Characterization of Novel Cell-Permeable, Small Molecule Adenylyl Cyclase Inhibitors for Future Development as Drugs to Treat FD/MAS”, Dr.
- “Single Cell Transcriptome Analysis of Skeletal Stem Cells Derived from FD/MAS Patients”, Dr.
- “Elucidating the Role of GNAS Mosaicism in Fibrous Dysplastic Lesions”, Dr.
Displasia Fibrosa y Embarazo
Muchas mujeres con DF/MAS tienen embarazos exitosos. No hay evidencia de que el embarazo haga que la DF empeore o aumente el dolor óseo. Las mujeres con DF en la columna, las costillas o la pelvis deben realizar un seguimiento con un obstetra especializado en embarazos de alto riesgo. Las mujeres que tienen antecedentes de pubertad precoz continúan teniendo quistes ováricos en la edad adulta. Esto puede derivar en sangrados irregulares e intensos. Las mujeres pueden tardar más en quedarse embarazadas.
Actividades Físicas
Los pacientes con DF deben usar el sentido común para decidir en qué actividades físicas van a participar. La DF craneofacial generalmente no altera las estructuras vitales del cráneo, incluyendo los vasos sanguíneos, el cerebro o las neuronas.
Manejo del Dolor
Cuando un paciente con DF sufre dolor, el primer paso es descartar causas ortopédicas, tales como fracturas, quistes del hueso, y problemas de la marcha. A los pacientes también se les debe revisar los niveles de fosfato, ya que niveles bajos pueden empeorar el dolor óseo. Si el dolor no está relacionado con estos factores, los primeros pasos en el tratamiento deben ser medidas conservadoras, incluyendo reposo, masaje, aplicación de frio y calor, y la ingesta de medicamentos como el ibuprofeno y el paracetamol.
Diagnóstico Diferencial
A pesar de que el patrón anatomopatológico es característico, se debe realizar el diagnóstico diferencial con múltiples entidades. El fibroma osificante es una verdadera neoplasia consistente en una proliferación de un tejido fibroso celular, que aparece bien delimitada o raramente encapsulada, que incluye hueso neoformado o calcificaciones esféricas. El tumor solo se presenta en los maxilares y huesos craneofaciales y tiene su inicio en tercera y cuarta década de la vida. Histológicamente, el componente fibroso muestra fibroblastos que no presentan atipia, escasas células multinucleadas gigantes, y ocasionales macrófagos espumosos. Las trabéculas óseas son finas, curvilíneas y aparecen inconexas, no hallándose bordeadas por un ribete de osteoblastos. Las características histológicas son similares a las de la DF por lo que la distinción hay que hacerla en base a los criterios clínicos, radiológicos, hallazgos quirúrgicos y características.
El querubismo es una enfermedad benigna, autosómica dominante, que consiste en una lesión de células gigantes de la mandíbula/maxilar que se presenta en niños entre 2-5 años como una tumefacción bilateral indolora, con progresión hasta la pubertad, cuando regresa de manera espontánea. Otras entidades que pueden confundirse clínicamente con la DF son el granuloma de células gigantes, el quiste óseo aneurismático, el cementoma gigante, el ameloblastoma o la displasia ósea, entre otros. Por ello es necesario un diagnóstico, además de clínico, basado en la imagen radiográfica (ya casi no utilizada) y más frecuentemente de TC, donde se describen varias formas (Schumberger, 1946):
- Pagetoide: expansión ósea con islas dispersas de formación ósea en campo de baja atenuación
- Esclerótica: apariencia homogénea con patrón en "vidrio esmerilado"
- Cística: lesión bien definida de baja atenuación con borde esclerótico en patrón de "cáscara de huevo"
Caso Clínico
Mujer de 14 años que en 2007 acude a nuestra consulta remitida por su odontólogo general para valorar posible tratamiento combinado ortodóncico-cirugía maxilofacial de la deformidad dento-facio-oclusal que presentaba la paciente. En el examen clínico inicial destacaba una facies microgénica con un perfil marcadamente convexo, mandíbula hipoplásica, incompetencia labial y sonrisa gingival por excesiva altura maxilar (fig. 1). La exploración intraoral reveló la presencia de una dentición mixta de 2.a fase con ausencia de 13, 14, 23 y 24. Maloclusión de clase II con 3mm de sobremordida y 6,5mm de resalte (fig. 2).
La radiografía panorámica, así como la oclusal palatina, mostraron múltiples inclusiones dentarias, así como desplazamiento hacia la línea media de 16, 26.
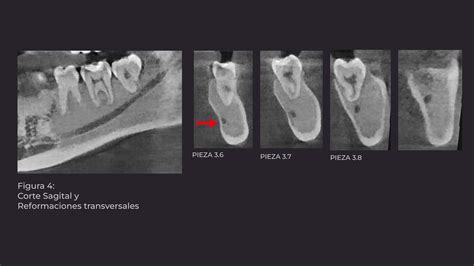
Se realizó Denta-Scan maxilar para valorar con más precisión la posición de los dientes incluidos10. Se observaron unos maxilares con aumento de densidad de forma generalizada, heterogénea, siguiendo un patrón radiológico en vidrio deslustrado. Al observar el maxilar se apreció una expansión grave de éste con ocupación parcial de los senos maxilares y retenciones dentarias múltiples con una situación muy alta y palatina de los caninos. El diagnóstico de presunción fue displasia fibrosa poliostótica (fig. 3).
Ante estos hallazgos, entre otras medidas, se decidió realizar una biopsia, que confirmó el patrón histológico de displasia fibrosa y se profundizó en la historia clínica de la paciente que inicialmente había referido estar en tratamiento por afección renal (nefrocalcinosis) con restricción dietética de calcio.
Con 2 años de edad, la paciente había sido diagnosticada de hipercalcemia secundaria a hiperparatoridismo primario con nefrocalcinosis secundaria. Inicialmente se planteó diagnóstico diferencial con síndrome de Williams, displasia cráneometafisaria, displasia ósea (tipo Rosenberg-Lohr) o condrodisplasia metafisaria tipo Jensen. Aunque cumplía alguno de los criterios, no era concluyente para ninguna de las entidades.
La paciente mejoró con tratamiento sintomático tanto de la calciuria y de la creatinina con corticoides (prednisona 10mg/48h) + retirada de productos lácteos.
Con 3 años de edad, la paciente inicia tratamiento con pamidronato (Aredia®) 1mg/kg/2 semanas durante 5 meses. Hubo mejoría analítica de todos los parámetros de metabolismo óseo, incluida una disminución de los valores de fosfatasa alcalina. Estos datos se confirmaron con una mejoría importante en la densitometría.
Por espacio de 3 años la paciente se encontraba clínicamente muy bien, con mejoría progresiva de la densitometría y con pauta alternante de alendronato (Fosamax®).
Con 8 años de edad se apreciaron lesiones óseas metafisarias que impresionaron de displasia ósea metafisaria. Se suspendieron los bisfosfonatos durante 6 meses para valorar su reintroducción en caso necesario. Tras este período, se reintrodujeron los bisfosfonatos por un período de 2 meses para tratamiento de hipercalcemia transitoria.
Finalmente, 6 meses antes de acudir a nuestra consulta, con 13 años de edad, se reintrodujo el tratamiento con alendronato (Fosamax®) 40mg/24h. En la actualidad la paciente ha suspendido el tratamiento de forma consensuada con su nefrólogo para disminuir el riesgo de necrosis maxilar como consecuencia del tratamiento combinado ortodóncico-quirúrgico, y dados los antecedentes de administración de bisfosfonatos orales e intravenosos de larga evolución.
En la actualidad la paciente se encuentra en tratamiento ortodóncico.
Tabla Resumen de la Displasia Fibrosa
| Característica | Descripción |
|---|---|
| Definición | Sustitución del tejido óseo normal por tejido conectivo fibroso. |
| Tipos | Monostótica (un hueso) y poliostótica (múltiples huesos). |
| Causa | Mutación en el gen GNAS1. |
| Síntomas | Dolor óseo, deformidades, fracturas, asimetría facial. |
| Tratamiento | Manejo del dolor, bifosfonatos, cirugía. |
| Pronóstico | Variable, depende de la extensión de la enfermedad. |