El Síndrome de Gardner es una variante fenotípica de la poliposis adenomatosa familiar (PAF), caracterizado por la presencia de pólipos intestinales, quistes epidérmicos y osteomas. Fue descrito en 1912, pero fue en 1953 que Gardner y Richards le dieron ubicación nosológica.
Su incidencia es de 1:8.000 a 1:16.000 nacidos vivos. Se transmite en forma autosómica dominante y exhibe alta penetrancia. El gen causal es el APC (adenomatous polypsosis coli) que codifica una proteína de 2.843 aminoácidos y mapea en el cromosoma 5q21. Dicho gen es la causa de diferentes expresiones fenotípicas: poliposis adenomatosa familiar, síndrome de Gardner y síndrome de Turcot (asocia poliposis colónica y tumores de sistema nervioso central).
El gen APC actúa como supresor tumoral. Al estar mutado, el gen APC favorecería el crecimiento tumoral, por desregulación de la cascada de la β catenina. Existe una correlación genotipo-fenotipo; de acuerdo con el lugar donde se produce la mutación varía la expresión clínica de la enfermedad.
Si la mutación se localiza en el tercio inicial o final del gen se asocia a una poliposis moderada, con manifestaciones tardías; si, en cambio, la mutación afecta a su parte central desarrolla enfermedad severa, con gran número de pólipos, asociada a manifestaciones extracolónicas a temprana edad.
Manifestaciones Cutáneas
Los quistes epidérmicos son la principal manifestación cutánea. Se observan en el 50 al 100% de los casos. En general aparecen en la infancia, son múltiples y se localizan en piernas, cara y cuero cabelludo (a diferencia de los quistes epidérmicos no sindrómicos que se localizan en dorso).
Su histología es similar a la de casos aislados y se caracteriza por una lesión quística, localizada en la dermis, compuesta por una pared de epitelio escamoso estratificado con presencia de granulosa, y una cavidad con contenido de queratina. En dos tercios de los pacientes se puede observar un aspecto similar a pilomatrixoma. Estos quistes tienen un curso benigno, y requieren exéresis sólo cuando presentan un problema estético, o cuando se rompen y provocan inflamación secundaria.
Los fibromas pueden aparecer en piel, tejido subcutáneo, mesenterio o retroperitoneo. Los tumores desmoides son característicos y se desarrollan en un 9% de los pacientes, se localizan en piel y tejidos blandos de pared abdominal anterior y en retroperitoneo. Se presentan en pacientes jóvenes con una edad promedio de 29,8 años; afectan con mayor frecuencia a las mujeres, con una relación mujer:varón de 3:1.
La histología evidencia un tumor poco circunscrito, compuesto en su mayoría por células fusiformes, con atipia ausente o mínima. Si bien no metastatizan, tienen un comportamiento agresivo e invasivo local. Tienden a recurrir luego de la extirpación quirúrgica.
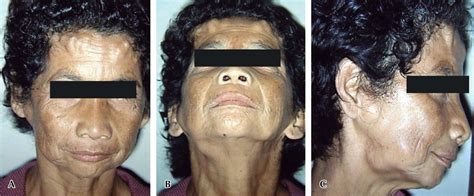
Otras alteraciones cutáneas observadas incluyen: lipomas (fig. 2), leiomiomas, neurofibromas y lesiones pigmentadas de piel.
Manifestaciones Extracutáneas
Las alteraciones oculares se presentan en el 90% de los pacientes, y son el primer hallazgo diagnóstico. Se caracterizan por la presencia de hipertrofias congénitas del epitelio pigmentado retiniano (CHRPE). Se presentan en forma bilateral, en mayor número y con pigmentación mixta (comparado con formas esporádicas) y se las considera un marcador específico de PAF. Es por este motivo que se recomienda un examen oftalmológico temprano.
Los osteomas son un hallazgo esencial del síndrome, y se observan al menos en el 50 al 75% de los casos; con incidencia en los huesos de la cara, en especial la mandíbula y el maxilar. Aparecen en la infancia, y preceden a la poliposis intestinal. Alcanzan su tamaño definitivo en algunos años, y luego continúan provocando cambios graduales aun en el adulto. Son neoplasias benignas, bien diferenciadas.
Pueden dividirse en dos tipos: centrales, que se desarrollan a partir del endostio, y periféricos, cuando se desarrollan del periostio. Para su detección se recomienda realizar una radiografía panorámica periódica de mandíbula, si bien la tomografía computarizada (TC) es un método más sensible de cribado. La escoliosis ha sido considerada parte del síndrome. También puede haber exostosis, endostosis y espesamiento cortical de los huesos.
Las anomalías dentales están presentes en un 18% de los casos. Son un signo temprano y se manifiestan con un espectro variable de alteraciones que incluyen: ausencia congénita de dientes, dientes supernumerarios e impactados en ambos maxilares, fusión de raíces dentales, multicaries, odontomas y quistes.
La poliposis intestinal es el rasgo más importante del síndrome; se presenta en el 100% de los pacientes. Se desarrolla en la pubertad, con una edad media de 22 años, y va aumentando en número con el tiempo. Se localiza en el colon y el recto, con frecuencia en los pliegues sigmoideo, esplénico y hepático. Pueden observarse con menor frecuencia en el intestino delgado y el estómago. Estos pólipos tienen un 100% de posibilidad de desarrollar adenocarcinoma; usualmente lo hacen en la tercera década de la vida.
Es por este motivo que se recomienda realizar estudios periódicos de sangre oculta en materia fecal, sigmoideoscopia, colonoscopia y endoscopia del tracto digestivo superior. La colectomía se recomienda ante la presencia de 30 o más pólipos, o cuando hay displasia o malignidad en un pólipo.
Si se preserva el recto, existe del 25 al 59% de posibilidad de desarrollar un carcinoma rectal tras la colectomía, motivo por el cual se recomienda también la resección completa de la mucosa rectal.
El segundo cáncer más común es el carcinoma periampular, que se observa en el 12% de los casos, desarrollado a partir de pólipos duodenales cercanos a la ampolla de Vater.
Se han descrito otros tumores asociados de páncreas, vejiga, tracto biliar, etc.
El cáncer de tiroides tiene mayor frecuencia con respecto a la población general y se produce a edades más tempranas. Generalmente son carcinomas papilares, que se desarrollan a una edad promedio de 23,6 años, más frecuente en mujeres. Por este motivo se recomienda un examen tiroideo y ultrasonografía periódica, y biopsia en lesiones mayores de 1 cm de diámetro.
A continuación, se presenta una tabla con los genes implicados en los cuadros de poliposis intestinal hereditaria:
| Gen | Síndrome |
|---|---|
| APC | Poliposis Adenomatosa Familiar (PAF), Síndrome de Gardner, Síndrome de Turcot |
| MUTYH | Poliposis asociada a MUTYH (MAP) |
| STK11/LKB1 | Síndrome de Peutz-Jeghers (SPJ) |
| PTEN | Síndrome de Cowden |
| SMAD4, BMPR1A | Poliposis Juvenil |
| ENG, ACVRL1 | Telangiectasia Hemorrágica Hereditaria |
| MSH2, MLH1, PMS2, MSH6 | Síndrome de Muir-Torre (variante de HNPCC) |